
Shielding Against AGE
June 5th, 2018The Crosslinkage Theory of Aging
by Ward Dean, M.D.
The Crosslinkage Theory of Aging was first proposed by Dr. Johan Bjorksten in 1941. Bjorksten believed that aging was caused by inter- and intra-molecular crosslinks in proteins, nucleic acids, and other vital macromolecules that caused them to gradually “stiffen” and lose their function.1
Bjorksten initially searched for enzymes capable of “dissolving” damaging crosslinks. But as he grew older he realized that he didn’t have enough years of life ahead of him to allow for the identification and isolation of these enzymes. Consequently, he shifted his line of research to a more immediately solvable approach: using chelating agents to remove toxic heavy metals, (especially aluminum) that were believed to be one cause of crosslinking. He hoped that by eliminating the crosslink-promoting tri-valent (three points of attachment) aluminum atoms, (which he believed displaced di-valent [two points of attachment] calcium atoms, he would reduce one of the major sources of crosslinking, and thereby “buy enough time” to solve the rest of the crosslinkage problem.2
Bjorksten ended his active research career in 1991 with one last publication that summarized his progress up to that point.3 Ironically, at about the time Bjorksten was retiring from his quest to unravel the crosslinkage problem, other scientists were “picking up the baton”- although they approached the problem from a slightly different direction.
Advanced Glycation End products of Aging (AGEs)
A characteristic of all long-lived proteins in the body is that as they age, they tum brown and become fluorescent (under UV light), become more crosslinked, less soluble, less elastic, and less digestible by enzymes. In 1965, Dr. H.B. Bensusan first proposed that it was a process known as the Maillard reaction that caused these changes. The Maillard reaction is named for the noted French scientist, Louis Camille Maillard (1912), who described the non-enzymatic chemical reactions between proteins and carbohydrates that cause cooked foods to turn brown. This time-honored bit of kitchen chemistry has been used by cooks for centuries to enhance flavor and transform plain foods into delicacies by adding flavor and color to recipes.
In 1985, Monnier, Kohn and Cerami provided further details of the role of the Maillard reaction as a major source of the age-dependent increase in browning, fluorescence and crosslinking of collagen and other tissues.4 They further developed the idea that it is the Maillard reaction that results in premature aging and degenerative diseases such as diabetes and heart disease. In this regard, many scientists think the human body may be viewed as a “low temperature oven” with a relatively long–approximately 75-100 year-“cooking cycle.”5
The Maillard reaction involves a chemical reaction (“condensation”) between a sugar (usually glucose) with a protein. This complex is known as a Schiff base. In the human body, this is a reversible reaction which reaches equilibrium (i.e., stabilizes) within several hours.
With continued exposure to the sugar, the Schiff base undergoes a “rearrangement” known as non-enzymatic glycosylation that results in a more stable, less reversible substance, known as an Amadori product. Again, in the human body, this process reaches equilibrium over several weeks. (Fig. 1)7
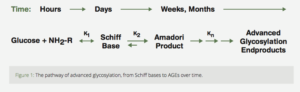
Figure 1: The pathway of advanced glycosylation, from Schiff bases to AGEs over time.
The Amadori product further degrades irreversibly into several highly reactive carbonyl (C=O) compounds. These reactive substances, called Advanced Glycation End products have been designated by the acronym AGE.5 AGE is a clever pun which reflects the proposed relationship of these reactive substances to aging and age-related diseases. AGEs can further react with other fats, proteins and nucleic acids to form largely indissoluble crosslinks. The age related accumulation of these AGE products has been demonstrated in many tissues of the body (Fig. 2).5
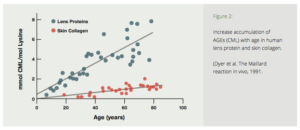
Figure 2: Increase accumulation of AGEs (CML) with age in human lens protein and skin collagen. (Dyer et al. The Maillard reaction in vivo, 1991.
Furthermore, during long-term hyperglycemia (elevated blood sugar), as in diabetics, glycation and AGE formation may increase up to four times as much! This explains why diabetics suffer the premature onset of a wide range of age-related complications including cataracts, retinopathy, neuropathy, nephropathy, atherosclerosis and osteoporosis. 6
Crosslinkage Theory Gets New Life
Bjorksten was a talented petroleum chemist. Had he been a food chemist instead, he may have appreciated this link between the Maillard Reaction and crosslinking much earlier and made even greater progress in developing preventive and therapeutic approaches to crosslink-induced aging. Through their insightful work in understanding this process, scientists like Brownlee, Cerami and Monnier provided a renewed impetus and a “rebirth” for the crosslinkage theory.6 Unfortunately, they did this with little attribution to Bjorksten, who had doggedly pursued this approach to aging for over 50 years.
Approaches to Preventing and Removing AGE-Induced Crosslinks
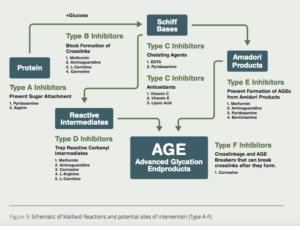
Scientists in the Departments of Biochemistry at the Universities of Kansas and South Carolina proposed a multi-pronged approach to inhibit the formation of AGEs (Fig. 3).7 Here are some of the most promising substances to use to inhibit/dissolve AGE-induced crosslinks.
Aminoguanidine (Pimagedine)
Aminoguanidine has been known to chemists for over 100 years. In 1986, aminoguanidine was found to prevent diabetes-induced arterial wall cross-linking in rats.8 Subsequent studies confirmed aminoguanidine’s ability to block the formation of AGEs and AGE-induced cross-linkages in collagen and other tissues (Fig. 4).9 Animal studies indicated that aminoguanidine prevented or delayed the onset of cataracts,10,11 inhibited atherosclerosis and myocardial stiffening,12-19 and protected against diabetic retinopathy,20-26 nephropathy20, 27-29 and neuropathy.30-31
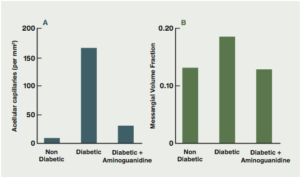
Figure 4: The effects of aminoguanidine on structural abnormalities of long-term diabetes in retina and glomerulus. (Brownlee, Diabetes Care, 1992).9
After nearly 15-years of in-vitro and in-vivo studies with rats, demonstrating the AGE-reducing properties of aminoguanidine, two large clinical trials were conducted to determine its efficacy in humans: Aminoguanidine Clinical Trial in Overt Nephropathy of Type 1 and Type II diabetics (“ACTION I” and “ACTION II”).32, 33
In the ACTION I trial, 690 diabetics with retinopathy and nephropathy were assigned to one of three groups: placebo; low dose (150 mg aminoguanidine twice daily); or high dose (300 mg aminoguanidine twice daily). The end-point of the study was the time it took for the serum creatinine to double from the baseline at entry into the trial. This would indicate a significant decline in renal function, a major complication of diabetes.
Serum creatinine doubled in only 20% of the pimagedine (aminoguanidine) group compared to 26% of the placebo group. Glomerular filtration rate (GFR, a sensitive indicator of kidney function) declined more slowly in the pimagedine-treated patients–and pimagedine reduced the 24-hour total urinary proteinuria. Surprisingly, the reduction was more pronounced in patients who received the low dose pimagedine. Pimagedine treatment was associated with a significant protection against doubling of creatinine. In addition, fewer Pimagedine-treated patients (10%) as compared with those receiving placebo (16%) experienced a three-step or greater progression of retinopathy; and treatment with low dose pimagedine was associated with a larger decrease in triglycerides and increase in high density lipoprotein cholesterol compared to placebo.
Mild adverse events included a transient flu-like syndrome between weeks 2 and 4 of treatment, which resolved spontaneously. Mild liver enzyme elevations were also occasionally noted, which were also self-limited and resolved spontaneously, regardless of whether pimagedine was continued or stopped. A mild to moderate anemia of unknown cause occurred in all study patients, (including placebo) but was more pronounced in those patients on pimagedine, especially during the first few weeks of treatment.
A more severe adverse effect, was crescentic glomerulonephritis which was observed in 3 patients in the high dose pimagedine group. Two of the three patients required maintenance dialysis. Significantly, glomerulonephritis was not observed in any of the 229 subjects receiving low dose pimagedine for an average of 2.5 years. It was this adverse effect that caused the trial’s FDA monitors to terminate the trial.
The authors of the report concluded that pimagedine produced a significant protective effect on the GFR compared with placebo; caused a significant decrease in proteinuria; was associated with a reduced progression of retinopathy; was associated with a reduction in the levels of total cholesterol and triglycerides and an increase in HDL cholesterol; and minimal adverse effects for those who received the lower, 300 mg/day dose.
“These effects are consistent with broad-spectrum activity of AGE formation on the pathogenesis of diabetic complications. The beneficial effects of pimagedine were apparent at the lower, 150 mg twice-daily dose, and this dose was well tolerated for a duration of exposure of up to 4.5 years. Toxicity observed with the higher dose of pimagedine was not noted with the lower dose.”32
Unfortunately, virtually every article that subsequently mentioned aminoguanidine (pimagedine) that I have seen referred only to the study’s early termination and exaggerated the side-effects, without mentioning the beneficial effects experienced by the patients on the low dose regimen. I think the backers of pimagedine, (Alteon Pharmaceuticals) were intimidated by the FDA and may have realized the difficulties they would encounter to have a generic substance like aminoguanidine approved as a profitable prescription drug, causing them to abandon any further evaluation and development. However, I think the study very authoritatively attested to the safety and benefits of low-dose (150-300 mg) aminoguanidine.
Metformin (Glucophage)
Metformin is an anti-diabetic biguanide that was derived from the herb, Goat’s rue (Galega officinalis). Biguanide drugs were recognized by Prof. Vladimir Dilman as early as the mid l970s as the most effective anti-aging drug in existence. Metformin is known primarily as an insulin receptor sensitizer, capable of normalizing blood sugar and insulin. However, Dilman demonstrated that biguanides restored receptor sensitivity for cortisol and other hormones, as well. Metformin has many other beneficial properties, including optimizing the lipid profile, reducing body fat, maintaining levels of growth hormone, stimulating immunity, and extending the maximum lifespan of experimental animals. I reviewed the anti-aging/ life-extending effects of Metformin in my article, “Metformin Update: Still the most effective anti-aging, life extension drug, with a broad range of benefits” in Aging Matters™ Magazine, No. 4, 2013.
Metformin is similar in structure to aminoguanidine (Fig. 5), suggesting that it may also have a potential effect on the inhibition of glycation reactions. Clinical studies in diabetics confirm metformin’s ability to prevent the formation of the potent AGE-precursor methylglyoxal (MG),35-38 and the AGEs carboxymethyl-lysine (CML)38,39 and pentosidine.39,40
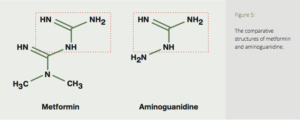
Figure 5: The comparative structures of metformin and aminoguanidine.
Pyridoxamine (and pyridoxal-5-phosphate [P-5-P])
Several compounds are commonly referred to as vitamin B6–pyridoxine, pyridoxal, pyridoxamine, and their respective 5`-phosphate forms. These are considered pyridoxine vitamers, and our body is capable of interconverting between them. Pyridoxamine is a metabolic precursor P-5-P that exerts antiglycative effects.7 The effect of pyridoxal, P-5-P, two forms of vitamin C (sodium ascorbate and dehydroascorbate [DHA]) and aminoguanidine were tested for their ability to prevent the non-enzymatic glycosylation (formation of AGEs) of bovine serum albumin (BSA) with radioactive-labeled glucose. P-5-P was exceeded only by aminoguanidine in its ability to inhibit AGE formation. (Fig. 6)41 Clinical studies of the P-5-P precursor pyridoxamine demonstrated it to be effective in reducing the increase of serum creatinine in diabetic patients with early nephropathy.42,43
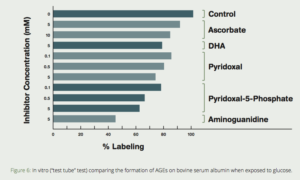
Figure 6: In vitro (“test tube” test) comparing the formation of AGEs on bovine serum albumin when exposed to glucose. Note the dramatic reduction of AGE formation when aminoguanidine or pyridoxal-5-phosphate are added. (Khatami, et al, Life Sciences, 1988)41
A recent study concluded that; “this antiglycative compound (pyridoxamine) exerts protective beneficial effects by reducing AGEs levels, leading to interferences with selective inflammatory and profibrotic signaling pathways.”44 Combining pyridoxamine with metformin and aminoguanidine may enhance their AGE-inhibiting actions even more.45
Vitamin B1 (Benfotiamine)
In their book, Life Extension, Durk Pearson and Sandy Shaw reported that thiamine was an effective crosslink inhibitor. They were, at that time, consuming two grams of thiamine each day in their personal anti-aging regimens. Benfotiamine is a lipid-soluble form of vitamin B1. It was found to be a potent inhibitor of glycation,46 inhibited the formation of AGEs and normalized nerve conduction velocity in diabetic rats,47 and blocked three major pathways of hyperglycemic damage and prevented diabetic retinopathy in rats.48
Scientists in Germany, aware that AGEs play a role in the development of endothelial dysfunction and vasculopathies, tested the ability of benfotiamine to reduce the AGE-precursor methylglyoxal (MG) and the AGE carboxymethyl-lysine (CML) in a group of patients with type II diabetes. Patients were given a cooked, high-AGE content test meal, and were then treated with 1000 mg of benfotiamine each day for three days and were then given another high-AGE test meal. Blood levels of MG, CML, and other tests of endothelial function, inflammation, and oxidative stress were measured before and after the test meals. The scientists found that benfotiamine prevents post-meal increases in circulating MG and CML levels in humans, and completely prevents micro- and macrovascular dysfunction caused by an AGE-rich test meal.49
Carnosine
Probably the leading proponent of the anti-aging effects of carnosine is Dr. Alan Hipkiss of the Division of Biomolecular Sciences, King’s College, London. Dr. Hipkiss first wrote of carnosine’s ability as a powerful crosslink inhibitor and AGE-breaker in 1994,50 and has subsequently produced a stream of papers describing its multiple uses to help control age-related glycation and associated diseases. Most recently, Dr. Hipkiss attributes methylglyoxal-induced AGEs as a cause of Parkinson’s disease, which may be alleviated by carnosine.51-52 Scientists in Turkey recently confirmed Hipkiss’ concepts by demonstrating age-related increases in the AGE-precursor methylglyoxal in rats, and the effectiveness of carnosine to reduce MG levels in the old rats.53 Carnosine’s AGE-inhibiting properties are the basis for the patented N-acetyl carnosine-containing eye drops (Can-C™), combined with an oral supplement of L-carnosine (Can-C+) to prevent and treat age-related cataracts and primary open-angle glaucoma.54
Carnitine
In 2007, scientists in India examined the anti-glycating effect of L-carnitine in rats fed a high-fructose diet to determine the potential of carnitine to inhibit in-vitro glycation. They found that the high-fructose diet caused hyperglycemia and glycation of hemoglobin and skin and tail tendon collagen; and that these effects were dramatically reduced in carnitine-fed rats. They concluded that carnitine not only has antiglycation effects but also provides evidence for the therapeutic use of carnitine in diabetes and associated complications.
In 2013, scientists in Japan found that diabetic patients with end-stage renal disease (ESRD) undergoing hemodialysis had higher levels of fluorescent AGEs in their skin compared to normal, non-diabetics, indicating that severe diabetics had higher levels of systemic glycation.
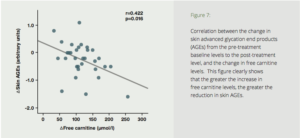
To test the effects of carnitine on AGEs in humans, seventy-two diabetics with ESRD were divided into two groups—a placebo group, and a test group treated with 1000 mg of carnitine daily for six months. Six months’ treatment with carnitine resulted in a dramatic reduction in skin auto-fluorescence (SAF) (Fig. 7). The carnitine treatment also resulted in improved endothelial and cardiac function, reduction of high-sensitivity C-reactive protein, and improved carotid pulsatility index. The authors concluded that suppression of AGE accumulation by L-carnitine may play a protective role against cardiovascular disease.
Figure 7: Correlation between the change in skin advanced glycation end products (AGEs) from the pre-treatment baseline levels to the post-treatment level, and the change in free carnitine levels. This figure clearly shows that the greater the increase in free carnitine levels, the greater the reduction in skin AGEs.
Conclusion
The venerable crosslinkage theory of aging bas clearly gained new respectability considering the advances in understanding of non-enzymatic glycation and the formation of AGEs and AGE-induced crosslinks. Although scientists have identified several substances that inhibit the formation of intermediates in the AGE-formation cascade (Fig. 3), the problem remains to find a truly effective cross-linkage breaker that will “undo the damage.” In the meantime, the most effective approach is to minimize the formation of advanced glycation end products (AGEs). This can be accomplished by consuming a low carbohydrate diet and exercising regularly, and adding a potent antiaging combination of AGE-inhibiting substances like aminoguanidine, metformin, carnosine, carnitine, benfotiamine and pyridoxamine or P-5-P.
References
- Bjorksten J. Aging: Present status of our chemical knowledge. J Am Geriatrics Soc, 1962, 10: 127.
- Bjorksten J, Possibilities and limitations of chelation as a means for life extension. Rejuvenation, 1980 VIII, No 3, 67-72.
- Bjorksten, J, Kleinsek DA. Synopsis of Prospective Longevity Research. 1991. Madison WI, JAB Publishing.
- Monnier VM, Kohn RP, Cerami A. Accelerated age-related browning of human collagen in diabetes mellitus. Proc Natl Acad Sci,1984,81: 583-587.
- Dyer DG, Blackledge JA, Katz BM, Hull CJ, Adkisson HD, Thorpe SR, Lyons TJ, and Baynes JW. The Maillard reaction in vivo. Z Ernahrungswiss, 1991, 30: 29-45.
- Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med, 1988, 318: 1315-132 l.
- Khalifah RG, Baynes JW, and Hudson BG. “Amadorins” Novel post-Amadori inhibitors of advanced glycation reactions. Biochem Biophys Res Com, 1999, 257: 251-258.
- Brownlee M, Vlassara H, Kooney A, Ulrich P, and Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science. 1986 Jun 27;232(4758):1629-32.
- Brownlee M. Glycation products and the pathogenesis of diabetic complications, Diabetes Care, 1992, 15: 1, 2, 1835-1843.
- Swamy-Mruthinti S, Green K, and Abraham EC. Inhibition of cataracts in moderately diabetic rats by aminoguanidine. Exp Eye Res, 1996, 62: 505-510.
- Yan H, Guo Y, Zhang J, Ding Z, Ha W, and Harding JJ. Effect of carnosine, aminoguanidine, and aspirin drops on the prevention of cataracts in diabetic rats. Mol Vis, 2008; 14, 2282-2291.
- Huijberts MSP, Wolffenbuttel BHR, Struijker-Boudler HAJ, Crijns FRL, AC, Nieuwenhuijzen Krese, Poitevin P, and Levy BI. Aminoguanidine treatment increases elasticity and decreases fluid filtration of large arteries from diabetic rats. J. Clin. Invest. Volume 92, September 1993, 1407-1411.
- Norton GR, Candy G, Woodiwiss AJ. Aminoguanidine prevents the decreased myocardial compliance produced by streptozotocin-induced diabetes mellitus in rats. Circulation. 1996; 93:1905-1912.
- Li YM, Steffes M, Donnelly T, Liu C, Fuh H, Basgen J, Bucala R, and Vlassara H. Prevention of cardiovascular and renal pathology of aging by the advanced glycation inhibitor aminoguanidine. Proc Natl Acad Sci USA. 1996 Apr 30; 93(9): 3902–3907.
- Corman B, Duriez M, Poitevin P, Heudes D, Bruneval P, Tedgult A, and Levy BI. Aminoguanidine prevents age-related arterial stiffening and cardiac hypertrophy. Proc. Natl. Acad. Sci. USA Vol. 95, pp. 1301-1306, February 1998.
- Chang KC, Hsu KL, Peng YI, Lee FC, and Tseng YZ. Aminoguanidine prevents age-related aortic stiffening in Fisher 344 rats: aortic impedance analysis. Br J Pharmacol. 2003, Sep; 140(1): 107-114.
- Chang KC, Hsu KL, Chou TF, Lo HM, and Tseng YZ. Aminoguanidine prevents age-related deterioration in left ventricular-arterial coupling in Fisher 344 rats. Br J Pharmacol. 2004 Aug; 142(7): 1099-1104.
- Chan V, Hoey A, and Brown L. Improved cardiovascular function with aminoguanidine in DOCA-salt hypertensive rats. Br J Pharmacol. 2006 Aug; 148(7): 902–908.
- Chang KC, Hsu KL, Chou TF, Lo HM, Tseng YZ. Aminoguanidine prevents arterial stiffening and cardiac hypertrophy in streptozotocin-induced diabetes in rats. Br J Pharmacolology. (2006) 147, 944-950.
- Soliman M. Preservation of myocardial contractile function by Aminoguanidine, a nitric oxide synthase inhibitors, in a rat model of hemorrhagic shock. Pak J Med Sci. 2013 Nov-Dec; 29(6): 1415–1419.
- El-Shazly AHM, Mahmoud AM, and Darwish NS. Potential prophylactic role of aminoguanidine in diabetic retinopathy and nephropathy in experimental animals. Acta Pharm 59 (2009) 67-73.
- Hammes HP, Martin S, Federlin K, Geisen K, and Brownlee M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc. Natl. Acad. Sci. USA, December 1991, Vol. 88, pp. 11555-11558.
- Roufail E, Soulis T, Boel E, Cooper ME, and Rees S. Depletion of nitric oxide synthase- containing neurons in the diabetic retina: reversal by aminoguanidine. Diabetologia (1998) 41: 1419-1425.
- Raskin P, Cattran D, Williams M, Wuerth JP, and Cartwright K. Pimagedine reduces progression of retinopathy and lowers lipid levels in patients with type 1 diabetes mellitus. J Am Soc Neph, 1999: 10 (PROGRAM AND ABSTR ISSUE): 179A.
- Mishra A, and Newman EA. Aminoguanidine reverses the loss of functional hyperemia in a rat model of diabetic retinopathy. Front Neuroenergetics. 2011, 3: 10.
- Luo D, Fan Y, Xu X. The effects of aminoguanidine on retinopathy in STZ-induced diabetic rats. Bioorg Med Chem Lett. 2012 Jul 1;22(13):4386-90.
- Carr BC, Emigh CE, Bennett LD, Pansick AD, Birch DG, and Nguyen C. Towards a treatment for diabetic retinopathy: Intravitreal toxicity and preclinical safety evaluation of inducible nitric oxide synthase inhibitors. Retina, 2017 Jan: 37(1):22-31.
- Soulis T, Cooper ME, Wanes D, Bucala R, and Jerums G. Effects of aminoguanidine in preventing experimental diabetic nephropathy are related to the duration of treatment. Kidney International, Vol 50 (1996), pp. 627—634.
- Forbes J, Soulis T, Tballas V, Panagiotopoulos S, Lon,g D, Vasan, et al. Renoprotective effects of a novel inhibitor of advanced glycation. Diabetologia, 2001, 44: I , !08 -114.
- Abdel-Rahman E, and Bolton WK. Pimagedine: a novel therapy for diabetic nephropathy. Expert Opin Investig Drugs. 2002; 11:566-74.
- Zimmerman GA, Meistrell M, Bloom O, Cockroft KM, Bianchi M, Risucci D, Broome J, Farmer P, Cerami A, Vlassara H, and Tracey KJ. Neurotoxicity of advanced glycation endproducts during focal stroke and neuroprotective effects of aminoguanidine. Proc Natl Acad Sci USA, 92, 3744-378, Apr 1995.
- Cockroft K. Meistrell J, Zimmerman G, Risucci O, Bloom O, Cerami A, and Tracey K. Cerebroprotective effects of aminoguanidine in a rodent model of stroke. Stroke, 1996, 27: 1393-1398.
- Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, Foiles PG, Freedman BI, Raskin P, Ratner RE, Spinowitz BS, Whittier FC, and Wuerth JP; ACTION I Investigator Group. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004 Jan-Feb;24(1):32-40. Epub 2003 Dec 17.
- Freedman BI, Wuerth JP, Cartwright K, Bain RP, Dippe S, Hershon K, Mooradian AD, Spinowitz BS. Design and baseline characteristics for the aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION II). Control Clin Trials. 1999 Oct;20(5):493-510.
- Beisswenger P, and Ruggiero-Lopez D. Metformin inhibition of glycation processes, Diabetes & Metabolism, 29, 4, Sept 2003, 6103.
- Beisswenger PJ, Howell SK, Touchette AD, Lal S, Szwergold BS. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes. 1999 Jan;48(1):198-202.
- Kiho T, Kato M, Usui S, Hirano K. Effect of buformin and metformin on formation of advanced glycation end products by methylglyoxal. Clin Chim Acta, 2005 Aug; 358(1-2):139-45.
- Kender Z, Fleming T, Kopf S, Torzsa P, Grolmusz V, Herzig S, Schleicher E, Rácz K, Reismann P, Nawroth PP. Effect of metformin on methylglyoxal metabolism in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2014 May; 122(5):316-9.
- Kinsky OR, Hargraves TL, Anumol T, Jacobsen NE, Dai J, Snyder SA, Monks TJ, and Lau S. Metformin Scavenges Methylglyoxal To Form a Novel Imidazolinone Metabolite in Humans. Chem Res Toxicol. 2016 Feb 15; 29(2): 227–234.
- Haddad M, Knani I, Bouzidi H, Berriche O, Hammami M, and Kerkeni M. Plasma levels of pentosidine, carboxymethyl-lysine, soluble receptor for advanced glycation end products, and metabolic syndrome: The Metformin effect. Dis Markers. 2016; 6248264.
- Chakraborty A, Chowdhury S, and Bhattacharyya M. Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients. Diabetes Res Clin Pract. 2011 Jul; 93(1):56-62.
- Khatami M, Suldan Z, David I, Li W, and Rockey H. Inhibitory effects of pyridoxal 5-phosphate, ascorbate and aminoguanidine on nonenzymatic glycosylation. Life Sciences, 1988; 43: 1725-1731.
- Williams ME, Bolton WK, Khalifah RG, Degenhardt TP, Schotzinger RJ, McGill JB. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol. 2007; 27(6):605-14.
- Lewis EJ, Greene T, Spitalewiz S, Blumenthal S, Berl T, Hunsicker LG, Pohl MA, Rohde RD, Raz I, Yerushalmy Y, Yagil Y, Herskovits T, Atkins RC, Reutens AT, Packham DK, Lewis JB; Collaborative Study Group. Pyridorin in type 2 diabetic nephropathy. J Am Soc Nephrol. 2012 Jan; 23(1):131-6.
- Chiazza F, Cento AS, Collotta D, Nigro D, Rosa G, Baratta F, Bitonto V, Cutrin JC, Aragno M, MastrocolaR, and M. Collino M. Protective Effects of Pyridoxamine Supplementation in the Early Stages of Diet-Induced Kidney Dysfunction. Biomed Res Int. 2017; 2682861.
- Ahmad S, Shahab U, Baig MH, Khan MS, Khan MS, Srivastava AK, and Saeed M. Inhibitory effect of metformin and pyridoxamine in the formation of early, intermediate and advanced glycation end-products. Plos One, 2013; 8(9).
- Brownlee M. Lilly Lecture 1993. Glycation and diabetic complications. Diabetes. 1994 Jun;43(6):836-41.
- Stracke H, Hammes HP, Werkmann D, Mavrakis K, Bitsch I, Netzel M, Geyer J, Köpcke W, Sauerland C, Bretzel RG, Federlin KF. Efficacy of benfotiamine versus thiamine on function and glycation products of peripheral nerves in diabetic rats. Exp Clin Endocrinol Diabetes. 2001; 109 (6): 330-6.
- Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q, Lin J, Bierhaus A, Nawroth P, Hannak D, Neumaier M, Bergfeld R, Giardino I, Brownlee M. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med. 2003 Mar; 9(3): 294-9.
- Stirban A, Negrean M, Stratmann B, Gawlowski T, Horstmann T, Götting C, Kleesiek K, Mueller-Roesel M, Koschinsky T, Uribarri J, Vlassara H, and Tschoepe D. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in Advanced Glycation End Products in individuals with Type 2 Diabetes. Diabetes Care 2006 Sep; 29(9): 2064-2071.
- Hipkiss AR, Michaelis J, Syrris P, Kumar S, Lan Y. Carnosine protects protein against in vitro glycation and cross-linking. Biochem Soc Transactions, 1994, 22, 3995.
- Hipkiss AR. On the Relationship between Energy Metabolism, Proteostasis, Aging and Parkinson’s Disease: Possible Causative Role of Methylglyoxal and Alleviative Potential of Carnosine. Aging Dis. 2017 May; 8(3): 334–345.
- Hipkiss AR. Glycotoxins: Dietary and Metabolic Origins; Possible Amelioration of Neurotoxicity by Carnosine, with Special Reference to Parkinson’s Disease. Neurotox Res. 2018 Feb 7.
- Bingül İ, Yılmaz Z, Aydın AF, Çoban J, Doğru-Abbasoğlu S, Uysal M. Antiglycation and anti-oxidant efficiency of carnosine in the plasma and liver of aged rats. Geriatr Gerontol Int. 2017 Dec;17(12):2610-2614.
- Babizhayev MA. Biomarkers and special features of oxidative stress in the anterior segment of the eye linked to lens cataract and the trabecular meshwork injury in primary open-angle glaucoma: challenges of dual combination therapy with N-acetylcarnosine lubricant eye drops and oral formulation of nonhydrolyzed carnosine. Fundam Clin Pharmacol. 2012 Feb;26(1):86-117.
- Rajasekar P, Anuradha CV. L-Carnitine inhibits protein glycation in vitro and in vivo: evidence for a role in diabetic management. Acta Diabetol. 2007 Jun;44(2):83-90.
- Fukami K, Yamagishi SI, Sakai K, Kaida Y, Adachi T, Ando R, and Okuda S.
Potential Inhibitory Effects of l-Carnitine Supplementation on Tissue Advanced Glycation End Products in Patients with Hemodialysis. Rejuvenation Res. 2013 Dec 1; 16(6): 460–466.




Download the latest FREE AGING MATTERS MAGAZINE
V3 issue 42 2023

- Advertisement -

